神经鞘瘤(Schwannomatosis, SWN),曾被称为施万细胞瘤,是一种通常为良性的肿瘤,起源于构成神经保护性髓鞘的施万细胞。过去,它被视为神经纤维瘤病的第三种类型,与I型和II型神经纤维瘤病并列。然而,由于其在遗传学和临床表现上与II型神经纤维瘤病(NF2)的显著相似性,最新的(2022年)诊断标准已将其细化,并纳入了NF2,形成了更精确的分类:
- NF2相关型神经鞘瘤
- SMARCB1相关型神经鞘瘤
- LZTR1相关型神经鞘瘤
- 染色体22q相关型神经鞘瘤
- 神经鞘瘤,非特指型(NOS)
- 神经鞘瘤,非其他分类(NEC)
不同类型的神经鞘瘤具有不同的临床肿瘤风险,如下图所示:
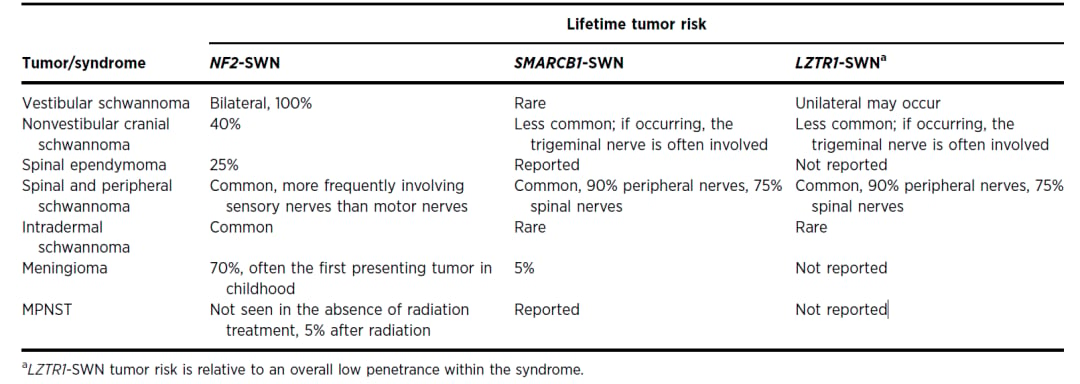
图1 NF2/SMARCB1/LZTR1相关型神经鞘瘤病罹患不同临床肿瘤的风险
理解这些分类及其背后的基因变异至关重要。例如,一项涉及12岁女童椎管神经鞘瘤的案例研究显示,通过基因检测发现其携带NF2基因胚系有害变异(丰度约50.71%),明确了其为NF2相关型神经鞘瘤,并提示存在较高的肿瘤遗传风险。这凸显了基因检测在神经鞘瘤精准诊断和风险评估中的核心价值。

图2 椎管神经鞘瘤患者检出的NF2基因胚系有害变异
一、深入了解NF2相关型神经鞘瘤
NF2相关型神经鞘瘤约占所有神经鞘瘤病例的3%,其根源在于NF2基因突变导致的II型神经纤维瘤病,这是一种常染色体显性遗传疾病。其临床特征表现为中枢神经系统(CNS)和外周神经系统的多发性肿瘤综合征。
主要临床表现包括:
- 中枢神经系统: 脑膜瘤、神经鞘瘤、胶质瘤、室管膜瘤等,其中双侧前庭神经鞘瘤是其标志性特征。
- 外周神经系统: 眼部异常(如白内障、视神经鞘脑膜瘤、视网膜错构瘤)和皮肤神经鞘瘤。
脊髓神经鞘瘤(髓外肿瘤)在这类患者中也较为常见,发生率可达55%-90%。
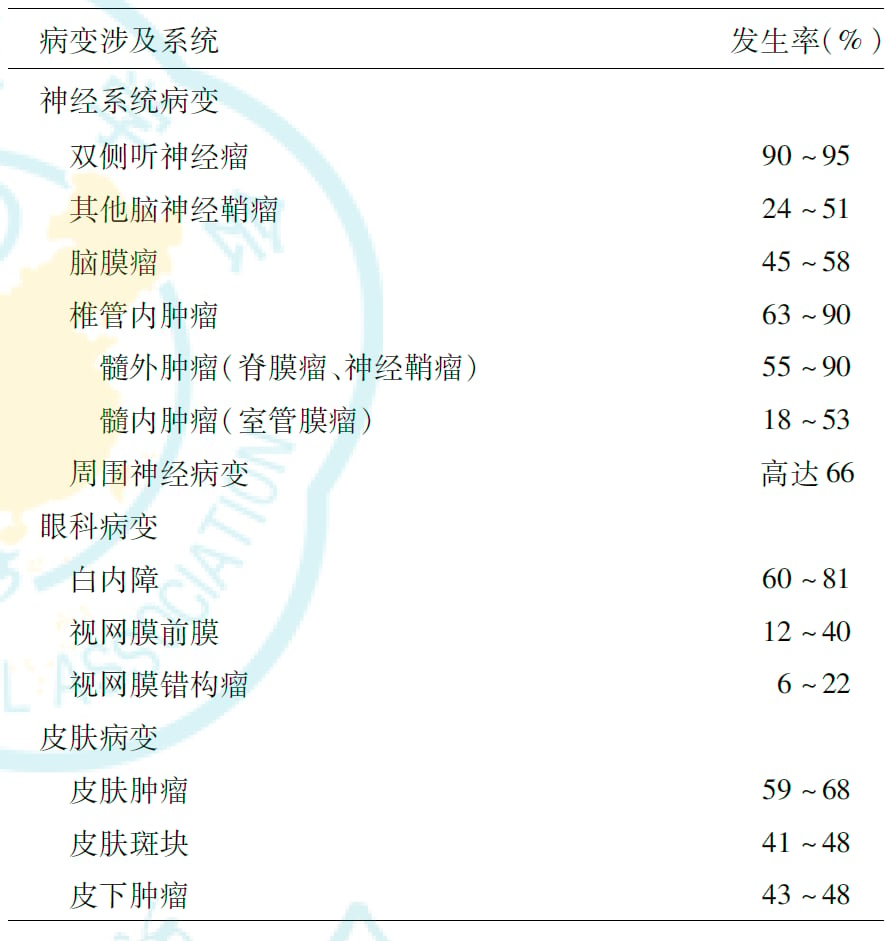
图3 II型神经纤维瘤病相关病变的发生率情况
NF2基因检测是诊断NF2相关型神经鞘瘤的关键辅助手段。根据2022年的标准,满足以下任一条件即可诊断:
- 确诊双侧前庭神经鞘瘤。
- 在不同解剖部位的两个NF2相关肿瘤(神经鞘瘤、脑膜瘤或室管膜瘤)中检测到相同的NF2基因突变。
- 满足以下组合:2个主要标准 或 1个主要标准 + 2个次要标准。
- 主要标准:①单侧前庭神经鞘瘤;②非同胞一级亲属患有NF2相关型神经鞘瘤;③两处或多处脑膜瘤;④正常组织(血液/唾液)检出NF2基因变异(若丰度显著低于50%,提示嵌合型)。
- 次要标准:①室管膜瘤、脑膜瘤或神经鞘瘤(同类可累积);②幼年期白内障、视网膜错构瘤、40岁以下视网膜前膜(同类不可累积)。

图4 NF2相关型神经鞘瘤的诊断标准(2022年)
NF2基因位于22号染色体长臂(22q12.2),包含17个外显子。其变异类型多样,包括错义、剪切、无义、移码突变及大片段缺失等。一项研究分析了57例确诊NF2患者,发现约49.1%存在NF2胚系变异,另有部分患者为嵌合型突变或未检测到突变,显示了检测的复杂性。
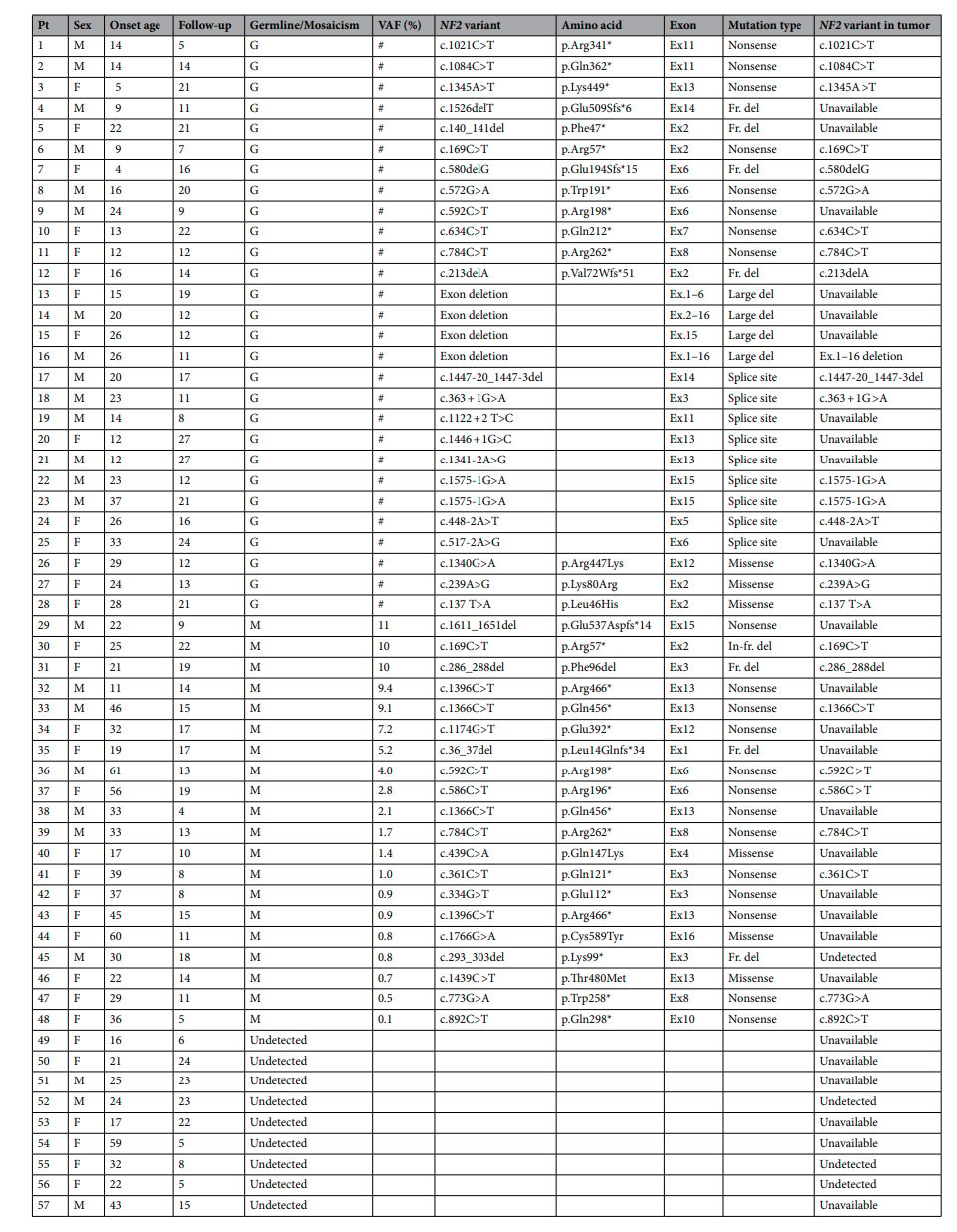
图6 57例确诊NF2患者的NF2基因胚系变异和嵌合变异位点
二、SMARCB1与LZTR1相关型神经鞘瘤
与NF2相关型相比,由SMARCB1或LZTR1基因胚系变异引起的神经鞘瘤较为少见(约占2%),且遗传外显率较低。
- SMARCB1相关型: 主要表现为周围神经鞘瘤,前庭神经鞘瘤不常见。
- LZTR1相关型: 临床表型也以周围神经鞘瘤为主,偶见单侧前庭神经鞘瘤。
基因检测同样是诊断这两种类型的关键。诊断标准包括:
- 至少一个病理确诊的神经鞘瘤(或混合性),且外周血/唾液检出相应的SMARCB1或LZTR1胚系有害变异(低丰度提示嵌合型)。
- 两个独立的神经鞘瘤病灶组织检出相同的SMARCB1或LZTR1有害变异。

图7 SMARCB1相关型和LZTR1相关型神经鞘瘤诊断标准
SMARCB1基因(位于22q11.2)和LZTR1基因(也位于22q11.2,更靠近着丝粒)的胚系变异位点已被多项研究报道,为基因检测提供了参考。
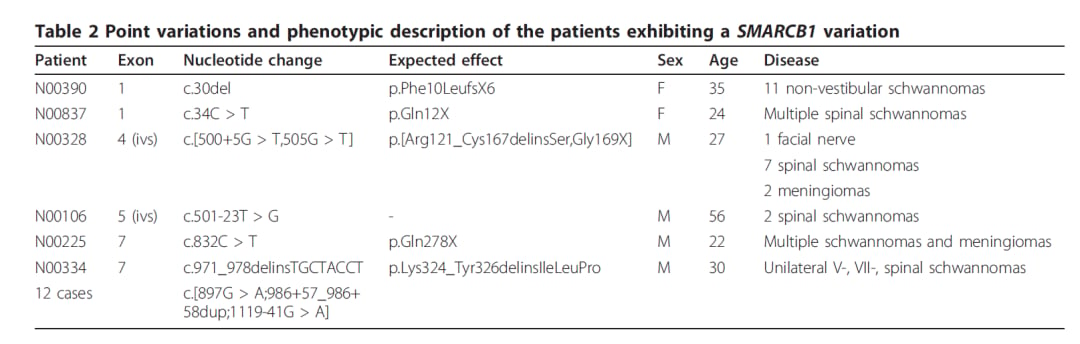
图8 56例非NF2型神经鞘瘤检出5例携带SMARCB1基因胚系有害变异患者
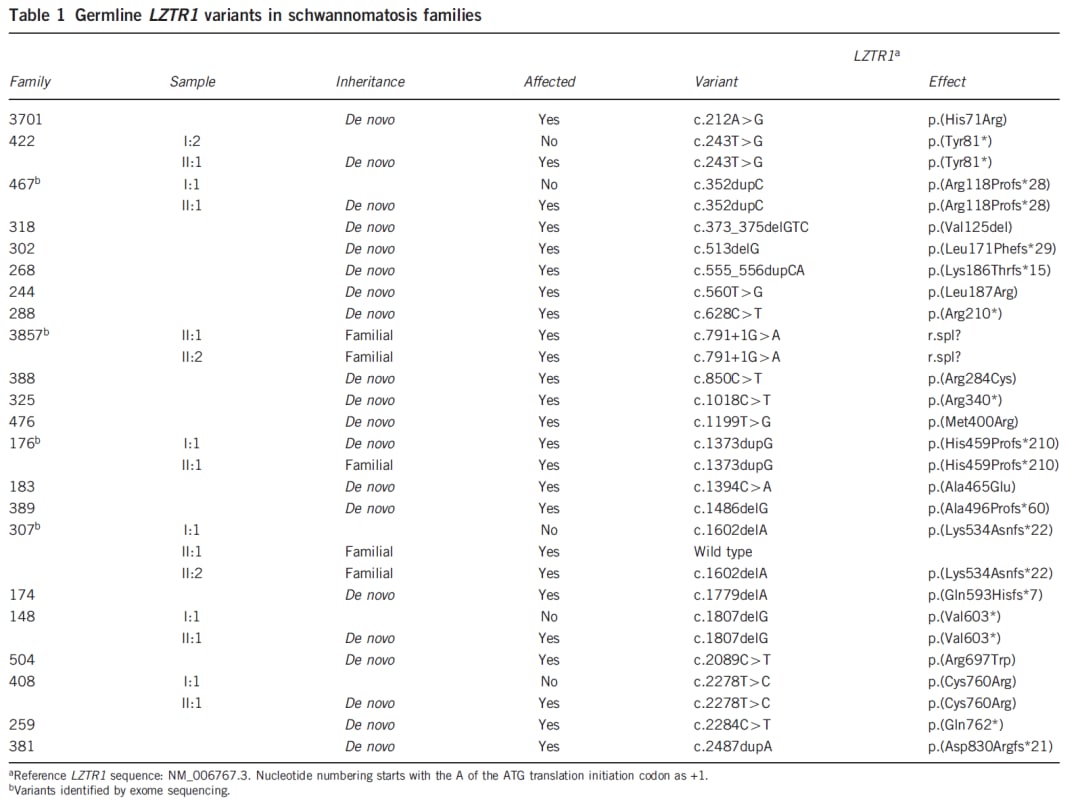
图9 LZTR1相关型神经鞘瘤患者的LZTR1基因胚系变异位点
三、其他类型的神经鞘瘤
除了上述由特定基因胚系变异定义的类型外,还存在其他几种神经鞘瘤分类:
- 染色体22q相关型: 不符合前三类标准,但在不同肿瘤病灶的相同22q位置检出杂合性缺失,且每个肿瘤有不同的NF2体细胞突变。
- 神经鞘瘤,NOS(非特指型): 未进行基因检测或结果未知,但影像学和病理学支持多发性非皮内神经鞘瘤。
- 神经鞘瘤,NEC(非其他分类): 经过充分基因检测,在正常组织和至少两个肿瘤病灶中均未发现已知相关的基因变异。

图10 其他类型神经鞘瘤的诊断标准
总结与展望
神经鞘瘤的精确诊断和分类高度依赖于基因检测。识别NF2、SMARCB1、LZTR1等基因的胚系变异,不仅有助于明确诊断、进行分子分型,更能评估患者及其家属的遗传风险。了解具体的基因突变类型对于评估预后和探索潜在的治疗方案至关重要。虽然目前针对这些特定基因突变的靶向药物仍在研究阶段,但精确的分子分型是未来个体化治疗和制定监测策略的基础。
如果您或家人被诊断为神经鞘瘤,或对相关的遗传风险、基因检测有疑问,可以考虑寻求专业的医疗建议。了解最新的诊疗信息和研究进展,对于管理此类疾病至关重要。如有进一步的疑问,MedFind AI问诊服务或可提供初步的资讯参考。
参考文献:
[1]Nagasaka, Shohei, and Ji Hoon Phi. “Genetic Basis and Clinical Management of Schwannomatosis.” Journal of Korean Neurosurgical Society, 10.3340/jkns.2025.0001. 6 Mar. 2025
[2]Perrino, Melissa R et al. “Update on Cancer and Central Nervous System Tumor Surveillance in Pediatric NF2-, SMARCB1-, and LZTR1-Related Schwannomatosis.” Clinical cancer research : an official journal of the American Association for Cancer Research, 10.1158/1078-0432.CCR-24-3278. 12 Feb. 2025
[3]中国抗癌协会神经肿瘤专业委员会.2型神经纤维瘤病神经系统肿瘤多学科协作诊疗策略中国专家共识[J].中华神经外科杂志, 2021, 37(7):6.
[4]Tamura, Ryota et al. “Historical Development of Diagnostic Criteria for NF2-related Schwannomatosis.” Neurologia medico-chirurgica vol. 64,8 (2024): 299-308.
[5]Teranishi, Yu et al. “Early prediction of functional prognosis in neurofibromatosis type 2 patients based on genotype-phenotype correlation with targeted deep sequencing.” Scientific reports vol. 12,1 9543. 9 Jun. 2022
[6]Perrino, Melissa R et al. “Update on Cancer and Central Nervous System Tumor Surveillance in Pediatric NF2-, SMARCB1-, and LZTR1-Related Schwannomatosis.” Clinical cancer research : an official journal of the American Association for Cancer Research, 10.1158/1078-0432.CCR-24-3278. 12 Feb. 2025
[7]Rousseau, Guillaume et al. “SMARCB1/INI1 germline mutations contribute to 10% of sporadic schwannomatosis.” BMC neurology vol. 11 9. 24 Jan. 2011
[8]张顺,刘丕楠,赵赋. 神经鞘瘤的分子遗传学研究进展[J]. 中华神经外科杂志,2017,33(9):966-969.
[9]Paganini, Irene et al. “Expanding the mutational spectrum of LZTR1 in schwannomatosis.” European journal of human genetics : EJHG vol. 23,7 (2015): 963-8.