错配修复蛋白缺失(dMMR)或微卫星高度不稳定(MSI-H)是结直肠癌、子宫内膜癌等多种实体瘤中非常关键的免疫治疗标志物。然而,当病理报告单上出现这些字眼时,许多患者和家属立刻会陷入“全家可能遗传癌症”的极度恐慌中。dMMR或MSI-H就一定等同于遗传性的林奇综合征吗?答案是否定的。在临床上,有多达50%的此类患者,既检测不到MMR基因的胚系致病突变,也没有MLH1启动子甲基化,这一群体在医学上被定义为“林奇样综合征(LLS)”。厘清两者的本质区别,对于患者制定精准的个体化治疗方案以及指导家族肿瘤筛查具有不可估量的生命价值。
林奇综合征 vs 林奇样综合征:核心特征深度对比
林奇综合征是由典型的错配修复(MMR)基因(如MLH1、MSH2、MSH6、PMS2)发生胚系致病性突变所致,具有显著的家族聚集性和早发性特征。而林奇样综合征则是一组高度异质性的疾病,其肿瘤组织同样表现出错配修复蛋白缺失和微卫星不稳定,但患者外周血中却检测不到经典的胚系突变。为了帮助患者更直观地理解,我们将三者的关键特征进行了多维度对比:
| 临床特征 | 林奇综合征 (LS) | 林奇样综合征 (LLS) | 散发性dMMR/MSI-H肿瘤 |
|---|---|---|---|
| 胚系突变 (外周血) | 存在MMR基因(MLH1/MSH2/MSH6/PMS2)致病性或疑似致病性突变 | 未检测到经典胚系致病突变(或仅有意义不明的变异VUS) | 无胚系突变 |
| 体细胞突变 (肿瘤组织) | 单等位基因突变+体细胞丢失 | 常见双体细胞突变(如MSH2/MSH6突变)或POLE基因等非MMR基因突变 | 无或不典型 |
| MLH1启动子甲基化 | 阴性 | 阴性 | 阳性(常伴有BRAF V600E突变) |
| 家族癌症遗传风险 | 极高,一级亲属需进行严格的早期多器官联合筛查 | 中等,亲属患癌风险低于林奇综合征,但高于普通人群 | 低,通常不具有家族遗传性 |
真实临床案例解析:林奇样综合征的异质性表型
由于林奇样综合征的定义在医学界仍存在不同的宽泛度,临床中极易出现漏诊或误诊。以下通过日常病理诊断中遇到的四个代表性临床案例,深度剖析不同基因变异背景下的复杂临床表现:
- 案例1:双体细胞突变导致的典型LLS。53岁直肠癌男性患者,免疫组化显示MSH2和MSH6蛋白表达缺失,为微卫星高度不稳定(MSI-H),且MLH1启动子甲基化为阴性。然而外周血基因检测未发现MMR致病性胚系突变(仅见PMS2基因的意义不明变异VUS),但在肿瘤组织中发现MSH2和MSH6基因的体细胞致病性突变。该病例最终确诊为由于肿瘤体细胞突变引起的林奇样综合征。
- 案例2:POLE基因突变驱动的LLS。62岁女性,同时确诊低分化子宫内膜样腺癌和结肠腺癌。子宫内膜癌组织表现为MSH2和MSH6蛋白缺失,外周血检测无MMR基因突变,但肿瘤NGS检测到POLE基因致病性突变。研究表明,POLE体细胞突变会引发超高突变负荷,从而诱导MMR蛋白表达缺失和微卫星不稳定,这也是LLS的一种重要隐匿机制。
- 案例3与案例4:被纠正的“非典型表型林奇综合征”。两名年轻女性患者分别表现为多原发癌(子宫内膜癌联合乳腺癌)或结肠多发息肉。由于最初其外周血MMR基因检测中的一些罕见突变被报告为意义不明的变异(VUS),曾被误诊为林奇样综合征。经过多学科专家深度评审并结合家系调查(如家族中存在严重的胃癌、结直肠癌等病史),最终确定其MLH1基因为疑似致病性突变,纠正诊断为非典型表型的经典林奇综合征。这提示我们在面对有明确家族史的患者时,诊断LLS必须慎之又慎。
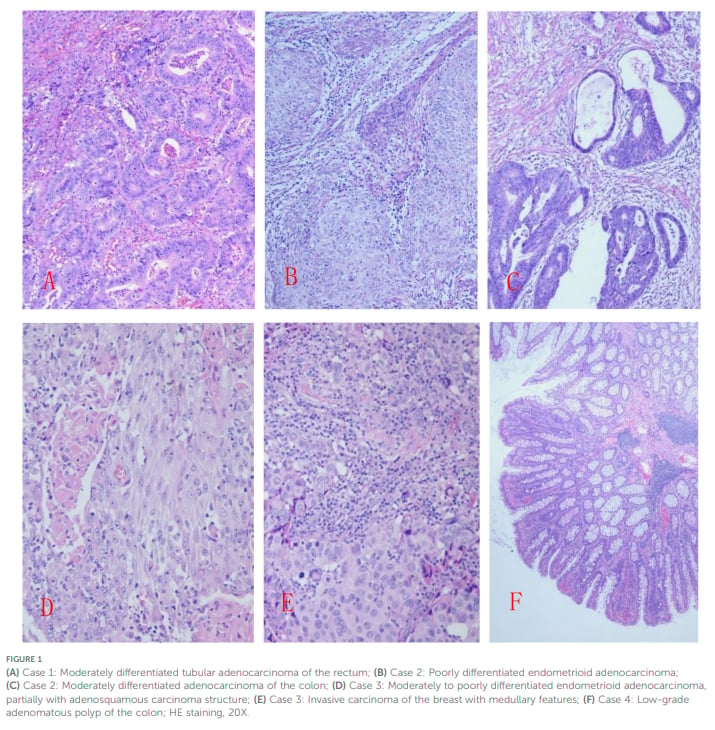
图1:临床疑似林奇样综合征患者各病灶的HE染色组织病理学特征(包含结直肠中分化腺癌、低分化子宫内膜样腺癌及伴髓样特征浸润性乳腺癌等)
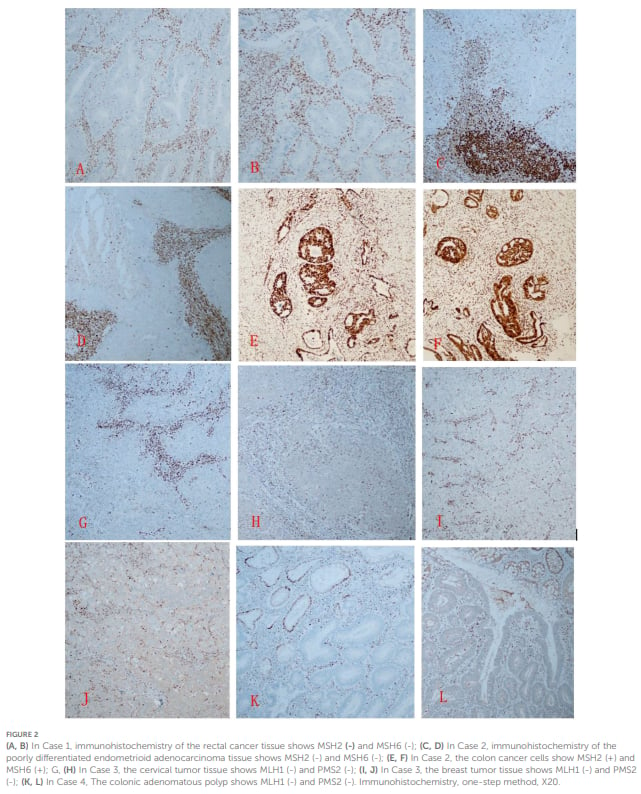
图2:各病例肿瘤组织中错配修复蛋白(IHC)表达检测,清晰展现不同突变类型下的蛋白缺失异质性
拨开迷雾:林奇样综合征背后的四大致病机制
为什么患者拥有经典的林奇表型却查不出胚系突变?医学界目前总结出了LLS背后的四种核心分子病理机制:
- 存在未被完全认识的“意义不明变异(VUS)”:部分突变虽然已被检出,但由于目前全球数据库中缺乏足够的临床证据,暂时被归类为VUS,这些变异未来极有可能被证实为致病突变。
- 隐匿性或复杂的结构变异:现有的常规二代测序(NGS)技术可能漏检一些内含子区域突变、染色体倒位或大片段拷贝数变异(CNVs)。
- 非MMR基因突变介导:包括POLE、POLD1、MUTYH等参与DNA复制与修复的其他关键基因发生体细胞或胚系突变,连锁导致了MMR系统的功能崩溃。
- 体细胞双突变与表观遗传事件:肿瘤局部的两个等位基因均发生后天体细胞突变,或是由于非经典的局部基因表观遗传沉默。
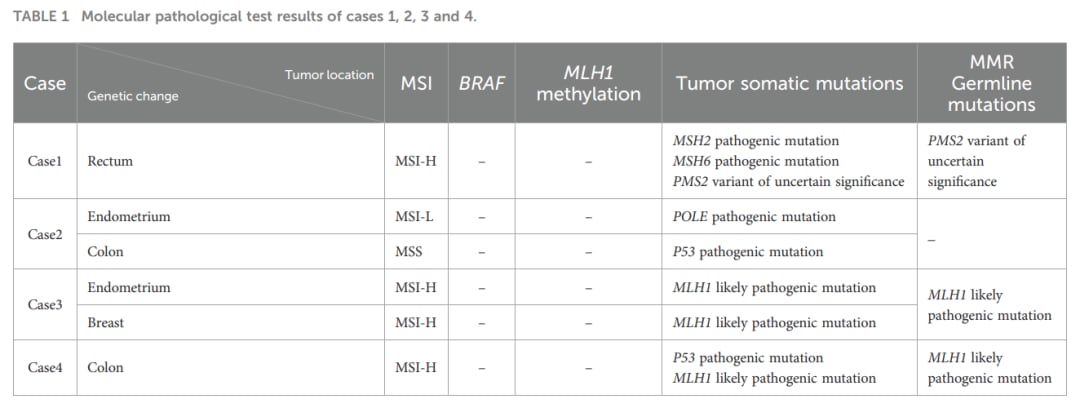
表1:各疑似病例的免疫组化、微卫星状态及启动子甲基化等详细分子病理学结果
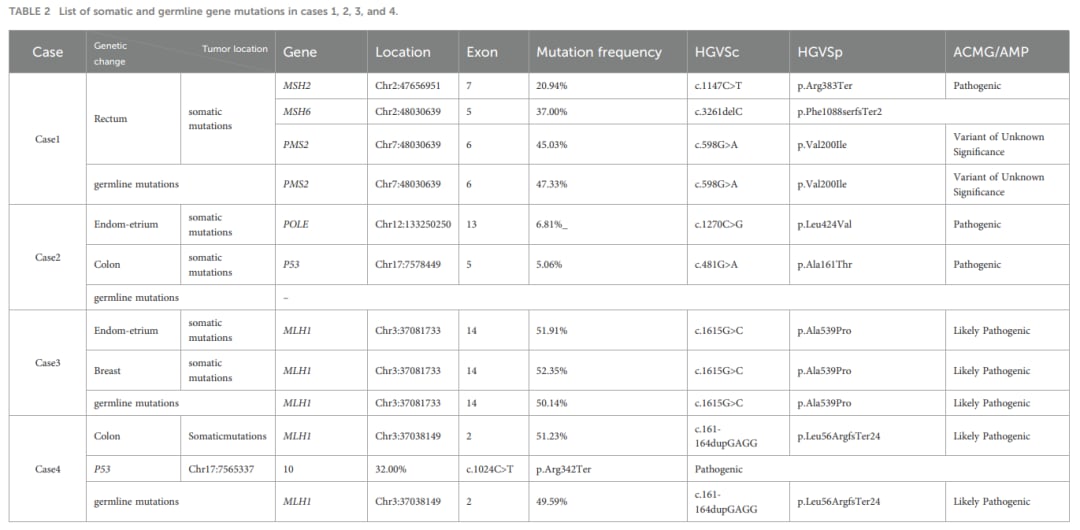
表2:各疑似病例通过二代测序(NGS)深度解码获取的胚系与体细胞突变基因变异详情
打破恐慌:dMMR/MSI-H患者的精准诊疗与用药指南
无论最终确诊的是林奇综合征还是林奇样综合征,对于已经发生肿瘤的患者来说,dMMR/MSI-H的核心表型都为其打开了一扇通往治愈希望的大门——免疫治疗。这类肿瘤微环境中有大量的淋巴细胞浸润,对免疫检查点抑制剂高度敏感。
1. 精准选择免疫检查点抑制剂
在临床上,针对dMMR/MSI-H特征的结直肠癌、子宫内膜癌等多实体瘤,以帕博利珠单抗(pembrolizumab)和纳武利尤单抗(nivolumab)为代表的PD-1抑制剂已经展现出令人瞩目的疗效,能显著延长患者的无进展生存期(PFS)和总生存期(OS)。哪怕是晚期患者,也有机会通过帕博利珠单抗等药物实现长期的带瘤生存甚至临床治愈。因此,精准区分病理类型不影响患者使用纳武利尤单抗等PD-1抑制剂的精准用药决策,但对于家庭层面的管理却至关重要。
2. 制定科学的家族防癌筛查计划
由于林奇样综合征患者的一级亲属累积患癌风险明显低于经典的林奇综合征家系,因此家族成员无需承受林奇综合征那样过于严苛和高频的筛查压力。然而,由于其患癌风险仍高于普通人群,家属仍应定期进行结肠镜检查及其他相关器官的健康监测,防患于未然。
MedFind守护抗癌之路:全球前沿药物与方案一站式可及
面对基因检测报告中复杂的“VUS变异”、“体细胞突变”以及繁多的免疫治疗选择,许多患者在求医路上常常感到迷茫与无助。部分前沿的高端多基因检测服务及全球最新获批的免疫靶向药物(如某些最新一代PD-1/PD-L1抑制剂及双抗疗法),在中国大陆的获批或引进仍存在一定的“时差”,给患者争取黄金治疗窗口期带来了极大挑战。
作为专注于消除医疗信息“边界”与“时差”的抗癌共享与互助平台,MedFind致力于为广大癌症患者提供全方位的支持。如果您手中有一份看不懂的基因检测报告,或者正面临耐药、寻找前沿临床用药的困境,MedFind的“AI辅助问诊与治疗方案解读”服务能够帮您迅速翻译、拆解复杂的医学学术报告,为您匹配全球权威的诊疗指南。同时,依托于健全的安全渠道,MedFind提供的“抗癌药品跨境直邮”服务,可帮助急需海外前沿药物的患者安全、合规、快速地获取全球最新获批药械,让生命无需等待,让希望触手可及。
【参考文献】
Cheng B, Liu S, Ding S, Quan L, Liu J, Xu L, Zhao H, Guo J and Sun S (2025) Pathological diagnosis experience and literature review of four cases suspected Lynch-like syndrome. Front. Oncol. 15:1608253. doi: 10.3389/fonc.2025.1608253