什么是林奇综合征?癌症的家族“魔咒”
林奇综合征(Lynch Syndrome, LS),又被称为遗传性非息肉病性结直肠癌(HNPCC),是临床上最常见的遗传性结直肠癌综合征。据统计,约有2%至4%的结直肠癌病例与其相关。这是一种常染色体显性遗传病,意味着子女有50%的几率从携带致病基因的父母一方继承该基因。其根源在于DNA错配修复(Mismatch Repair, MMR)系统中的关键基因发生了胚系突变,这些基因包括我们熟知的 MLH1、MSH2、MSH6、PMS2,以及一个名为上皮细胞黏附分子(EpCAM)的基因。
当MMR系统功能失常,细胞在分裂过程中产生的DNA复制错误就无法被及时纠正,导致基因组高度不稳定,这种情况在分子诊断上被称为“高度微卫星不稳定性(MSI-H)”。这种基因层面的不稳定极大地增加了携带者罹患多种癌症的风险,其中最为典型的就是结直肠癌。此外,林奇综合征还与子宫内膜癌、胃癌、卵巢癌、脑肿瘤等多种癌症的风险显著升高有关。不同基因的突变,其对应的癌症风险谱和严重程度也各不相同,这使得林奇综合征的管理极具挑战性。
一例罕见的家族病例:25岁确诊结肠癌
医学界最近报告了一例极为罕见的林奇综合征家族案例,为我们揭示了其背后复杂的遗传机制。该案例的先证者是一名年仅25岁的年轻男性,因右下腹持续疼痛一个月而就医。通过结肠镜检查,医生在他的升结肠发现了一个环形隆起的肿块,病理活检最终证实为腺癌。影像学检查也显示升结肠壁存在不对称增厚。幸运的是,经过及时的3D腹腔镜右半结肠切除术,术后病理分期为pT3N0M0(IIA期),属于中期但无远处转移。由于分期较早,他并未接受辅助化疗,并在长达56个月的随访中保持无病生存状态。
然而,这位年轻患者的背后,是一个触目惊心的癌症家族史。在他的家族谱系中,横跨五代共30名亲属里,竟有多达8人罹患癌症。其中包括他的父亲(29岁患直肠癌)、叔叔(32岁患结肠癌)、祖母(64岁患子宫内膜癌),以及其他亲属罹患的脑肿瘤、乳腺癌和胰腺癌。如此显著的家族聚集性和早发性特征,高度提示了林奇综合征的存在。

▲图1 LS家族的临床和分子检测
基因检测的惊人发现:四基因共突变与大片段缺失
为了揭开这个家族癌症频发的谜团,研究人员对先证者的肿瘤组织和家族成员的外周血进行了深入的基因检测,结果令人震惊。
肿瘤组织的独特发现
首先,对患者切除的肿瘤组织进行免疫组化(IHC)检测,结果显示 EpCAM、MSH2 和 MSH6 三种蛋白的表达缺失,而 MLH1 和 PMS2 蛋白表达正常。这一结果直接指向了 MSH2 通路的功能缺陷。进一步的二代测序(NGS)分析,在肿瘤组织中发现了一个前所未有的、跨越 EpCAM 基因外显子8-9和 MSH2 基因外显子1-16 的新型大片段缺失。这种大范围的基因结构变异极具破坏性,是导致MMR功能丧失的直接原因。同时,微卫星不稳定性(MSI)分析也证实肿瘤呈现出MSI-H状态,与林奇综合征的分子特征完全吻合。

▲表1 6例EpCAM和MSH2共突变的LS病例
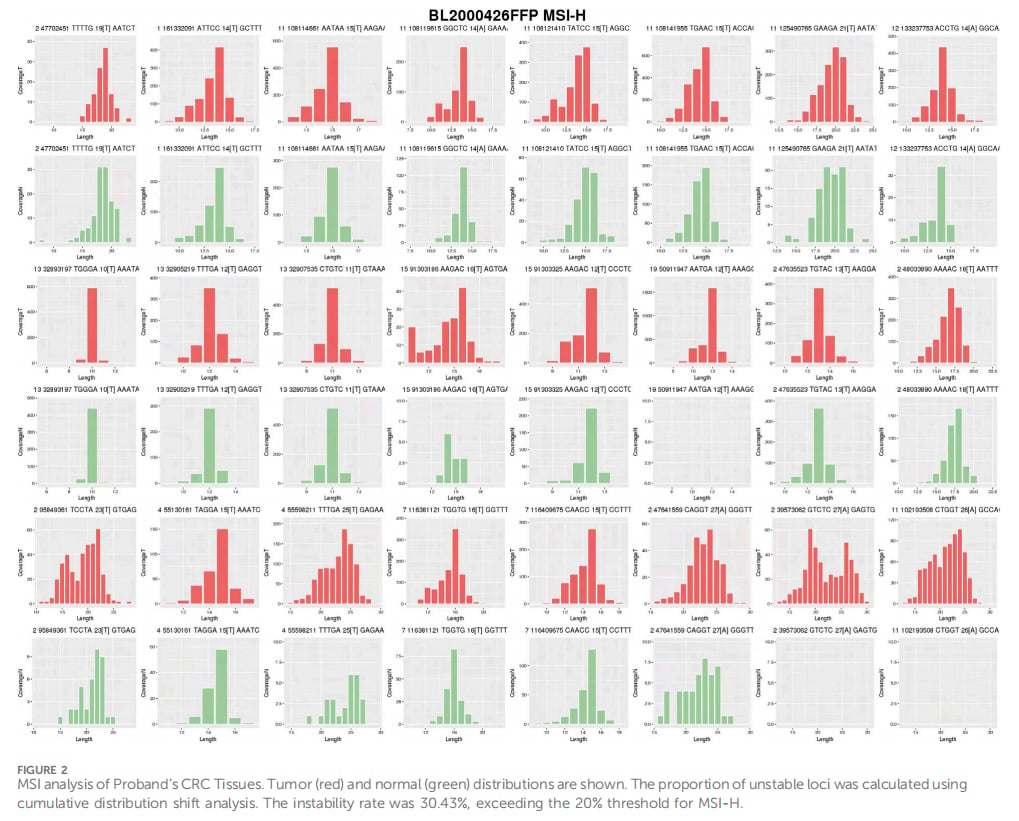
▲图2 先证者CRC组织MSI分析
家族成员的遗传图谱
更令人意外的是,当研究人员对包括先证者、其叔叔、姑姑以及年仅6岁的儿子在内的6名家族成员进行外周血全外显子测序(WES)时,发现他们所有人都携带了四种基因的共突变:EpCAM (c.344T>C)、MSH2 (c.2744A>G)、PMS2 (c.1408C>T) 和 APC (c.5465T>A)。APC基因是另一个著名的与结直肠癌相关的基因,其突变通常与家族性腺瘤性息肉病(FAP)有关。这种多基因共存的现象在林奇综合征中极为罕见,文献中从未有过EpCAM、MSH2和PMS2共突变的报道。
最关键的发现来自先证者年仅6岁的儿子。在他的外周血样本中,不仅检测到了上述四种点突变,还检测到了与他父亲肿瘤组织中完全相同的EpCAM-MSH2大片段缺失。这一发现强烈暗示了“胚系嵌合”现象的存在,即这种致病性的大片段缺失可能存在于生殖细胞中,并遗传给了下一代,这将大大增加其在极早期罹患癌症的风险。
共突变的临床意义:为何数据库显示“良性”?
这个案例最令人困惑的一点是,当研究人员查询公共基因数据库时,发现检测到的四种点突变大多被归类为“良性”或“意义未明(VUS)”。这与该家族严重的癌症史形成了鲜明对比。这揭示了当前基因检测解读的一大挑战:
- 单一突变的局限性: 公共数据库通常基于单个突变在普通人群中的频率和生物信息学预测来评估其致病性。然而,它们可能无法评估多个“低风险”突变共同存在时产生的协同效应或“上位性效应”。在本案例中,EpCAM、MSH2、PMS2和APC这四个基因分别在MMR通路和Wnt/β-连环蛋白通路中扮演重要角色,它们的共同改变可能相互作用,共同推动了肿瘤的发生。
- 预测工具的不足: 不同的生物信息学预测工具对同一突变的致病性预测结果常常不一致,甚至相互矛盾。例如,SIFT预测MSH2突变为“致病性”,而其他工具则认为是良性。这凸显了单纯依赖计算机预测的局限性,功能性实验验证至关重要。
- 结构变异的重要性: 真正驱动该家族癌症风险的,很可能是那个在常规数据库中难以检索的EpCAM-MSH2大片段缺失。这提醒我们,在进行遗传性肿瘤筛查时,不能仅仅关注点突变,还必须综合评估拷贝数变异等结构性改变。
面对如此复杂的基因检测结果,如何解读并制定下一步的健康管理计划?MedFind的AI问诊服务可以为您提供专业的分析和建议,帮助您理解复杂的报告,并与您的医生共同决策。
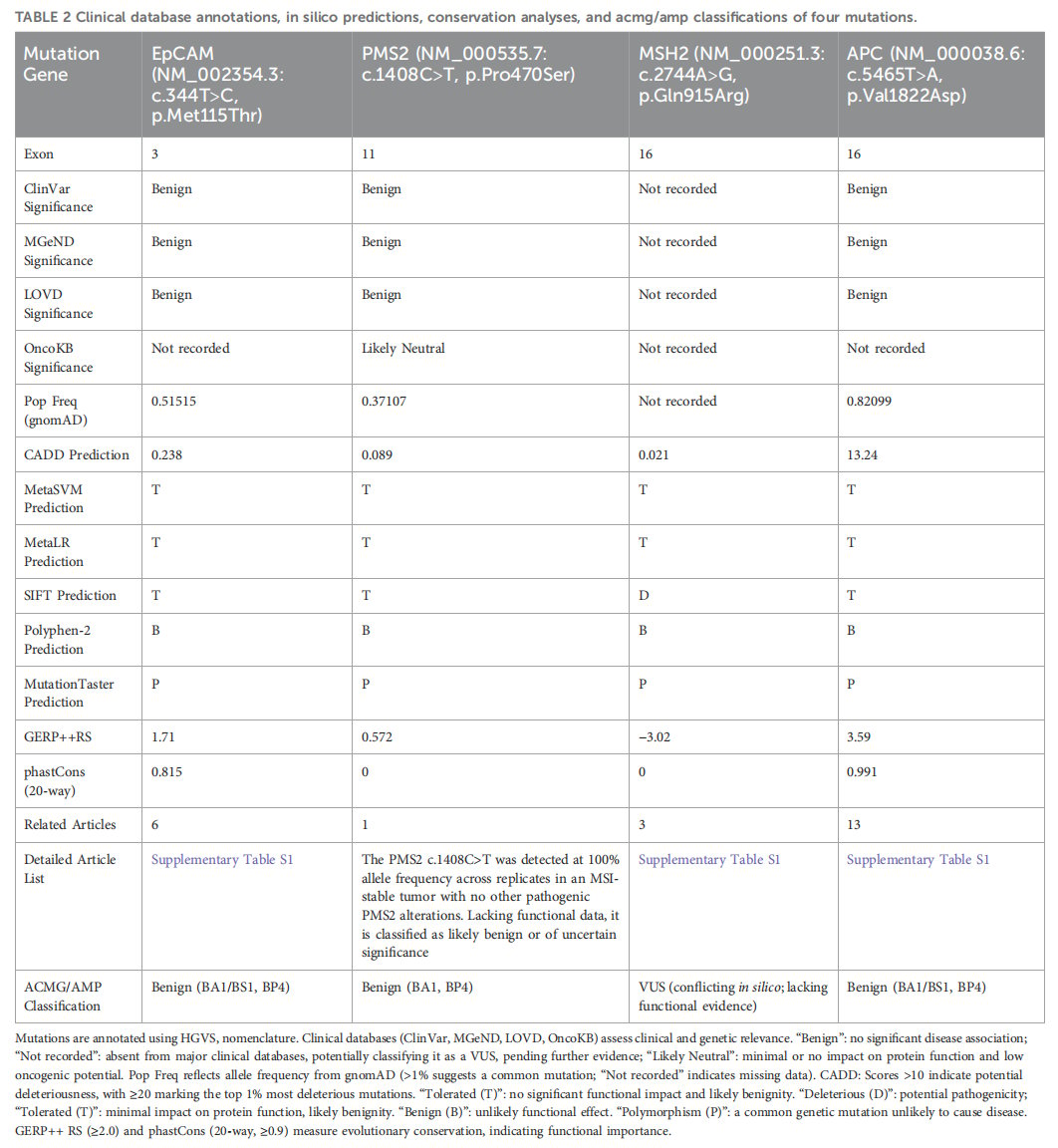
▲表2 四种突变的临床数据库注释、计算机预测、保守性分析和ACMG/AMP分类
对高风险家族的启示:早期筛查与积极监测
这个独特的家族案例为所有林奇综合征家族和其他遗传性癌症高风险人群敲响了警钟,并提供了宝贵的管理启示:
- 全面基因筛查的必要性: 对于有强烈癌症家族史的个体,特别是存在早发性癌症、多发性癌症的家族,应考虑进行更全面的基因检测,如全外显子测序(WES),而不仅仅是针对单个或几个常见基因的检测。
- 监测起始年龄需大幅提前: 根据现行指南,MMR基因突变携带者通常建议从20-25岁开始接受每一到两年一次的结肠镜检查。但在这个家族中,最早的结直肠癌患者年仅25岁。因此,对于该家族的携带者,监测起始年龄应大幅提前至20岁之前,甚至更早。
- 多癌种系统性监测: 林奇综合征的风险不局限于结直肠。该家族出现了高比例的脑肿瘤(25%),远高于普通LS家族。因此,携带者应接受包括神经系统评估(必要时进行脑部MRI)、女性妇科检查(经阴道超声、子宫内膜活检以筛查子宫内膜癌和卵巢癌)在内的多方位、系统性监测。
- 级联筛查至关重要: 一旦家族中确认了致病性突变,应对所有一级亲属进行“级联筛查”,以识别出所有无症状的携带者。这能帮助他们尽早进入监测计划,通过早期发现和干预,有效降低癌症的发病率和死亡率。
制定个性化的监测方案至关重要。获取更多关于林奇综合征及相关癌症的最新诊疗资讯,您可以访问MedFind抗癌资讯板块,我们为您提供前沿的医学信息。
结论与展望
总而言之,这个携带EpCAM、MSH2、PMS2和APC罕见共突变的林奇综合征家族,不仅拓宽了我们对该疾病遗传谱系的认知,也深刻揭示了在解读复杂遗传信息时面临的挑战。它强调了在评估遗传性癌症风险时,必须超越单个基因的局限,综合考虑点突变、大片段缺失以及多基因协同作用的可能。尽管许多问题仍待进一步的功能实验来验证,但这一案例无疑为临床实践提供了重要参考。
对于高风险家族而言,积极进行遗传咨询、接受全面的基因检测、并严格遵循个性化的早期筛查和监测方案,是有效对抗家族“癌症魔咒”的最有力武器。而对于已经确诊的患者,治疗方案的选择同样关键,包括错配修复缺陷(dMMR)状态对免疫治疗的指导意义等。MedFind致力于为患者提供全球前沿药物的代购和直邮服务,帮助患者第一时间获得最佳治疗选择。